İçindekiler
Tanım
PMM2 konjenital glikozilasyon bozukluğu (PMM2-CDG, konjenital glikozilasyon bozukluğu tip Ia olarak da bilinir) vücudun birçok parçasını etkileyen kalıtsal bir durumdur. PMM2-CDG ile bağlantılı problemlerin tipi ve şiddeti, etkilenen bireylerin arasında hatta bazen aynı ailenin üyeleri arasında çeşitli farklılıklar gösterir.
PMM2-CDG olan bireyler genellikle bebeklik boyunca bu durumun belirti ve semptomlarını gösterirler. Etkilenen bebekler; zayıf kas tonusu (hipotoni), çatlamış (tersine çevrilmiş) meme ucu, normal olmayan yağ dağılımı, aynı yöne bakmayan gözler (şaşılık), gelişimsel gecikme ve kilo kazanımı ve beklenen oranda büyüme eksikliğine (büyüme aksaklığı) sahiptirler.
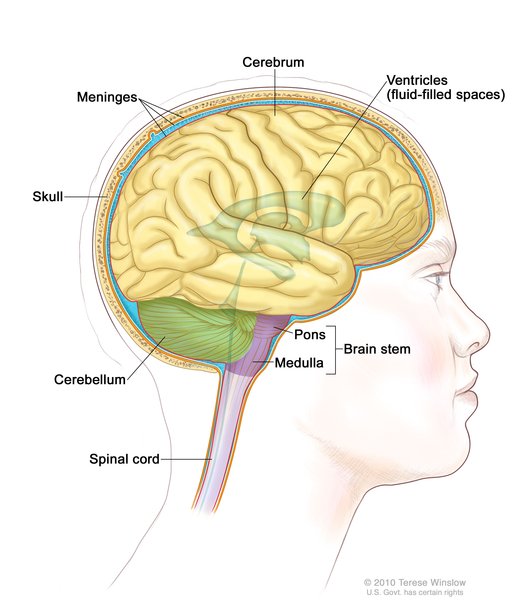
PMM2-CDG’ye sahip bebeklerde, beynin parçası olan hareketleri koordine eden çoğu zaman gelişmemiş olan beyincik (Şekil 1) görülür. Etkilenen bazı bireylerde yüksek alın, üçgen yüz, büyük kulaklar ve ince üst dudak olmak üzere karakteristik yüz özellikleri vardır.
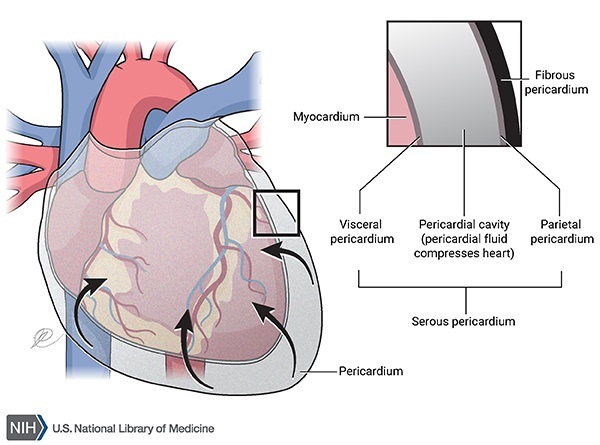
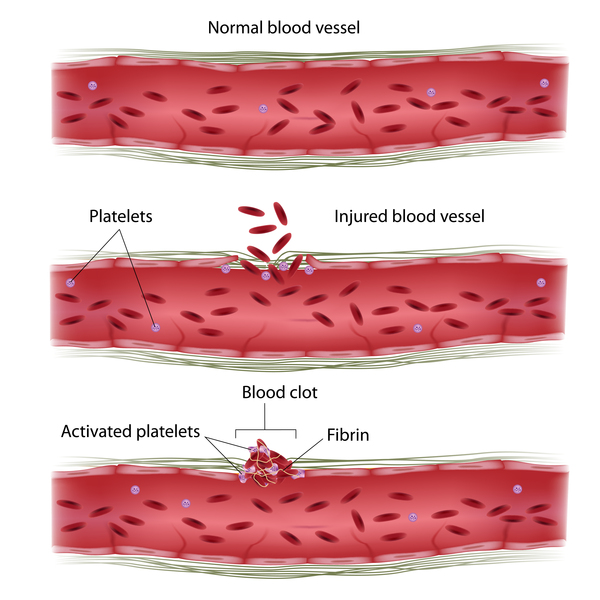
PMM2-CDG’ye sahip olan çocuklar ayrıca yüksek karaciğer fonksiyonu test sonuçları, kasılma nöbetleri, kalp etrafında sıvı birikimi (perikard efüzyonu) (Şekil 2) ve kan pıhtılaşması (Şekil 3) hastalıklarına sahip olabilir. Etkilenen bebeklerin yaklaşık olarak %20’si yaşamlarının ilk senesindeki çoklu organ yetmezliğine bağlı olarak hayatta kalamaz.
Şiddetli PMM2-CDG vakalarının birçoğu doğumdan önce vücutta aşırı sıvı birikmesi olan “hidrops fetalis” durumuyla ilişkilendirilir. Hidrops fetalise sahip olan bebeklerin çoğu ölü doğar veya doğumdan hemen sonra ölür. PMM2-CDG’ye sahip olan bebeklik döneminde hayatta kalabilen insanlarda hafif zihinsel yetersizlik ve bazılarının bağımsızca yürüyemediği görülür. Etkilenen bireylerde ayrıca aşırı enerji eksikliği (halsizlik) dahil olmak üzere felç benzeri nöbetler ve geçici felç gözlenebilir. Bu nöbetlerden kurtulma genellikle birkaç haftadan birkaç aya kadar olan zaman diliminde gerçekleşir.
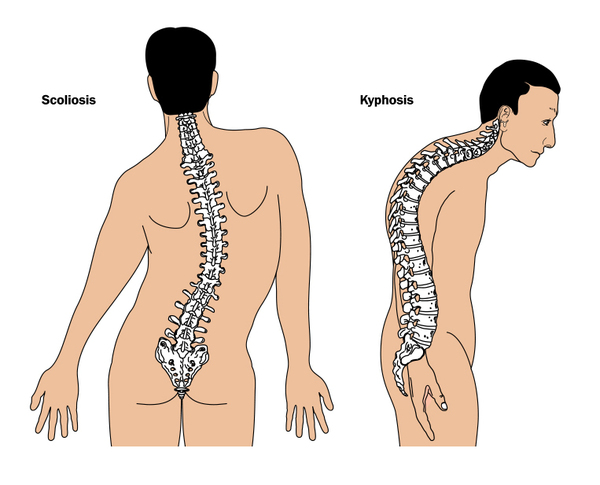
Yetişkinlik veya ergenlik dönemi boyunca PMM2-CDG’si olan bireylerde duyularda azalma, kollarda ve bacaklarda güçsüzlük (periferik nöropati), anormal omurga eğriliği (kifoskolyoz) (Şekil 4), kas koordinasyonunda bozulmalar (ataksi) ve eklem bozulmaları (kontraktürler) görülür.
Etkilenen bazı bireylerde, görme kaybına yol açan ve pigmenter retinopati denilen bir göz hastalığı vardır. PMM2-CDG’ye sahip olan kadınlarda, cinsel gelişimi yönlendiren hormonların üretimini etkileyen “hipergonadotropik hipogonadizm” görülür. Sonuç olarak PMM2-CDG’ye sahip olan kadınlar ergenliğe girmez. Etkilenen erkekler normal ergenlik geçirirler fakat genellikle küçük testislere sahip olurlar.
Sıklık
Dünya çapında PMM2-CDG’ye sahip olan 800’den fazla birey tespit edilmiştir.
Nedenler
PMM2-CDG, PMM2 genindeki mutasyondan kaynaklanmaktadır. Bu gen, fosfomannomutaz 2 (PMM2) denilen enzimin yapımı için bilgi sağlamaktadır. PMM2 enzimi bir grup şeker moleküllerini (oligosakkaritler) proteinlere bağlayan glikozilasyon denilen sürece dahil olmaktadır. Glikozilasyon proteinleri modifiye ederek daha çeşitli fonksiyonlar göstermelerini sağlar.
PMM2 genindeki mutasyon, aktivitesi azalmış ve anormal PMM2 enziminin üretimine yol açar. Fonksiyonlarını düzgün bir biçimde yerine getiren PMM2 enziminin yokluğunda glikozilasyon normal bir şekilde ilerleyemez. Sonuç olarak, yanlış oligosakkaritler üretilir ve proteinlere bağlanır. PMM2-CDG’nin çok çeşitli belirti ve semptomları olası birçok doku ve organda anormal bir biçimde glikolize edilmiş proteinlerin üretiminden ortaya çıkmaktadır.
Kalıtım Modeli
Bu durum otozomal çekinik modelle kalıtılır, bu da her hücredeki genin her iki kopyasında da mutasyon olduğu anlamına gelmektedir. Otozomal çekinik durumu olan bireyin ebeveynlerinin her birinde mutasyonlu genin bir kopyası bulunur fakat genellikle bu durumun belirti ve semptomlarını göstermezler.
Bu Hastalığın Diğer İsimleri
- Karbonhidrat eksikliği olan glikoprotein sendromu tip Ia
- CDG Ia
- CDG1a
- CDGS1a
- Glikosilasyon tip Ia’nın konjenital bozukluğu
- Jaeken Sendromu
- Fosfomannomutaz 2 eksikliği
- PMM eksikliği
- PMM2-CDG
Kaynak: https://medlineplus.gov/genetics/condition/pmm2-congenital-disorder-of-glycosylation/
Görsel Kaynak: https://medlineplus.gov/genetics/condition/pmm2-congenital-disorder-of-glycosylation/#synonyms
Editör: Hazal Kalsın Demir