

İçindekiler
COL1A2 Geni: Kollajen Tip I Alfa 2 Zinciri
Normal Fonksiyonu
COL1A2 geni, tip I kollajen denilen büyük bir molekülün bir kısmının üretilmesi emrini verir. Kolajenler, kıkırdak, kemik, tendon, cilt ve gözün beyaz kısmı (sklera) dahil olmak üzere vücuttaki birçok dokuyu güçlendiren ve destekleyen protein ailesidir. Tip I kollajen insan vücudunda en bol bulunan kollajen şeklidir.
Pro-α2 (I) zinciri olarak adlandırılan bir tip I kollajen bileşeni, COL1A2 geninden üretilir. Kolajenler, her biri üç zincirden oluşan ip benzeri prokollajen moleküllerinden meydana oluşur. Tip I kollajen (COL1A1 geninden üretilen) iki pro-α1 (I) zincirinden ve bir pro-α2 (I) zincirinden oluşur.
Üç sarmallı prokollajen molekülleri, olgun kollajen oluşturmak için hücre dışındaki enzimler tarafından işlenir. Kolajen molekülleri daha sonra kendilerini hücreler arasındaki boşluklarda birbirleriyle dengeli etkileşimler (çapraz bağlantılar) oluşturan uzun, ince liflere düzenler. Çapraz bağlantılar, çok güçlü tip I kollajen liflerinin oluşumu ile sonuçlanır.
Genetik Değişikliklerle İlgili Sağlık Koşulları
Ehlers-Danlos Sendromu
COL1A2 genindeki çeşitli mutasyonlar, artrochalasia tipi olarak bilinen bir çeşit Ehlers-Danlos sendromuna neden olabilir. Ehlers-Danlos sendromu, cildi, kemikleri, kan damarlarını ve diğer birçok organ ve dokuyu destekleyen bağ dokularını etkileyen bir hastalık grubudur. Artrochalasia tipi, alışılmadık derecede geniş bir eklem hareket aralığı (hipermobilite) ve doğumda her iki kalçanın çıkığı ile karakterizedir. Her hücrede COL1A2 geninin bir kopyasını etkileyen genetik değişiklikler, kritik bir segmenti olmayan bir pro-α2 (I) zincirinin üretimine yol açar. Bu segmentin yokluğu, pro a2 (I) zincirlerinin olgun tip I kollajen moleküllerinde birleştirilmesine ve işlenmesine engel olur. Bu değişiklikler temel olarak cilt, kemikler ve tendonlar gibi tip I kollajen bakımından zengin dokuları etkiler.
Nadiren, her hücrede COL1A2 geninin her iki kopyasındaki mutasyonlar, kalp-valvüler tip olarak tarif edilen bir Ehlers-Danlos sendromu formunda insanlarda bildirilmiştir. Bu nadir durum kalp kapakçıklarındaki anormallikler, oldukça esnek (elastik) cilt ve eklem hipermobilitesi ile karakterizedir. Hastalığın bu formuna neden olan mutasyonlar, hücrelerin normal pro-a2 (I) zincirlerini üretmesini önler. Sonuç olarak, cilt ve diğer dokulardaki tip I kollajen fibrilleri doğru şekilde birleştirilemez. Anormal kollajen bağ dokusunu zayıflatır ve bu durumun belirti ve semptomlarına neden olur.
Osteogenez İmperfekta
Çoğu COL1A2 gen mutasyonu, tip II, III ve IV dahil olmak üzere ciddi osteogenez imperfekta formlarına neden olur. Bu koşullara sahip kişilerin, genellikle hafif travmadan veya belirgin bir nedeni olmayan kolayca kırılan kemikleri vardır. COL1A2 genindeki mutasyonlar zaman zaman bu hastalığın en hafif şekli olan osteogenez imperfekta tip I’e neden olur.
Bazı COL1A2 mutasyonları, kritik bölgeleri olmayan bir pro-α2 (I) zincirine yol açan gen parçalarını siler. Diğer genetik değişiklikler, pro-a2 (I) zincirindeki protein yapı bloklarının (amino asitler) dizilimini değiştirir, genellikle amino asit glisinini farklı bir amino asitle değiştirir. Bazı durumlarda, amino asit ikameleri, kollajen moleküllerinin birleştirilmesini engelleyen protein zincirinin bir ucunu (C-terminus olarak adlandırılır) değiştirir. Bu COL1A2 mutasyonları normal tip I kollajen üretimini önler. Anormal kollajen gelişmekte olan kemiklere ve diğer bağ dokularına dahil edildiğinde, şiddetli osteogenezis imperfekta formlarıyla ilişkili ciddi tıbbi sorunlara neden olur.
Diğer Bozukluklar
Bazı COL1A2 mutasyonlarına sahip kişiler, hem osteogenez imperfekta hem de Ehlers-Danlos sendromunun (yukarıda açıklandığı gibi) belirtilerini ve semptomlarını gösterir. Bu mutasyonlar, genin büyük bir kısmının kopyalarını, pro-a2 (I) zincirinin önemli bir bölümünün silinmesini ve pro-a2 (I) zincirinin anormal şekilde kısaltılmış bir versiyonuyla sonuçlanan genetik değişiklikleri içerir. COL1A2 genindeki mutasyonlar, bağ dokusunu zayıflatan ve bu iki durumun karakteristik özelliklerine yol açan tip I kollajen fibrillerinin yapısını değiştirir.
Kromozomal Konum
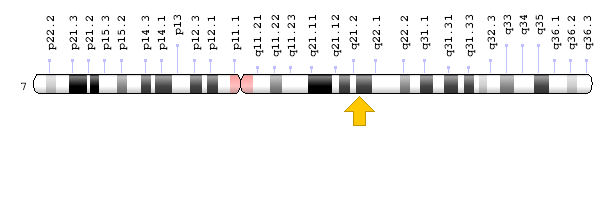
Sitogenetik Konum: 7q21.3, 21.3 konumundaki kromozom 7‘nin uzun (q) kolu
Moleküler Konum: kromozom 7 üzerindeki 94,394,561 -94,431,232 baz çiftleri
Bu Genin Diğer İsimleri
- alpha 2 collagen type I
- CO1A2_İNSAN
- collagen I, alpha-2 polypeptide
- collagen of skin, tendon and bone, alpha-2 chain
- collagen type I alpha 2
- collagen, type I, alpha 2
Kaynak: https://ghr.nlm.nih.gov/gene/COL1A2
Görsel Kaynak: https://nauka.tass.ru/nauka/6815452
Editör: Meryem Melisa KAR
