
İçindekiler
DPYD Geni: Dihidropirimidin Dehidrojenaz
Normal Fonksiyonu
DPYD geni, timin ve urasil moleküllerinin ihtiyaç duyulmadığında parçalanmasında yer alan “dihidropirimidin dehidrojenaz” olarak isimlendirilen bir enzimin yapısını oluşturan proteinin sentezi için gerekli emirleri verir. Timin ve urasil , bir tür nükleotid olan pirimidinlerdir. Nükleotitler, DNA’nın, kimyasal kuzeni RNA’nın ve hücrede GTP ve ATP gibi enerji kaynağı olarak hizmet eden moleküllerin yapı taşlarıdır.
Dihidropirimidin dehidrojenaz, pirimidinlerin parçalanmasının ilk aşamasında yer alan enzimdir. Bu enzim timini 5,6-dihidrotimine dönüştürür ve urasili 5,6-dihidrourasil denilen başka bir moleküle dönüştürür. Pirimidinler parçalandığında açığa çıkan moleküller vücut tarafından ya atılır ya da diğer hücresel işlemlerde kullanılır.
Genetik Değişiklikler Sonucu Oluşan Sağlık Durumları
Dihidropirimidin Dehidrojenaz Eksikliği
Dihidropirimidin dehidrojenaz eksikliği olan kişilerin DPYD geninde 50’den fazla mutasyonun bulunduğu tespit edilmiştir. DPYD gen mutasyonlarının, urasil ve timinin bazlarının parçalanmasını tamamen engellediği ya da enzim aktivitesi azaltığı, parçalanamayan timin ve urasil molekülleri idrar, kan ile beyni ve omuriliği çevreleyen sıvı (beyin omurilik sıvısı) içinde aşırı miktarlara birikimine neden olmaktadır. Fazla timin ve urasilin, dihidropirimidin dehidrojenaz eksikliği olan insanları etkileyen bazı spesifik nörolojik problemlerle ilişkisi henüz belirsizliğini korumaktadır.
İlginçtir ki; DPYD geninde oluşan mutasyonların, “kapesitabin” ve “5-florourasil” gibi pirimidinlere benzer yapılara sahip kanser ilaçlarının parçalanmasına da müdahale ettiği keşfedilmiştir. Bu keşiften sonra, dihidropirimidin dehidrojenaz eksikliği olan bazı kişilerde ortaya çıkan ciddi reaksiyonların, kanser tedavisinde kullanılan bazı ilaçlarının Dehidronejenaz enziminin eksikliğinden dolayı parçalalanmaması sonucu dhidroprimidinin vücutta birikmesinin sebep olduğu anlaşılmıştır.
Koloboma
Koloboma, gözün anormal gelişiminden kaynaklanır. Doğumdan önceki gelişimin ikinci ayında, gözün yapılarını oluşturmak için optik fissür (koroid fissür veya embriyonik fissür olarak da bilinir) olarak isimlendirilen bir dikiş kapanır. Optik fissür tamamen kapanmadığında, sonuç bir kolobomadır.
Gelişim sırasında göz küresinin dibinde optik çatlak oluştuğu için gözün alt yarısında kolobomlar ortaya çıkar. Gözdeki kolobomdan etkilenen belirli yapı, optik fissürün kapanamayan kısmına bağlıdır.
Koloboma, gözün erken gelişiminde yer alan birçok gendeki değişikliklerden kaynaklanabilir, bunların çoğu tanımlanmamıştır. Durum ayrıca bir veya daha fazla geni etkileyen bir kromozomal anormallikten kaynaklanabilir.
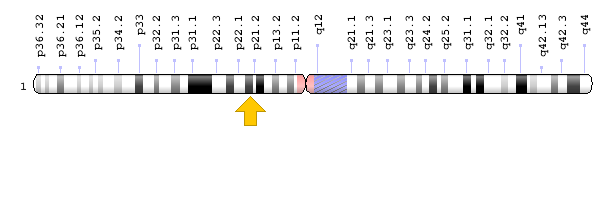
Kromozomal Konum
- Sitogenetik Konum: 1p21.3, kromozom 1’in kısa (p) kolunun 21.3 konumunda
- Moleküler Konum: Kromozom 1’de 97,077,743 ile 97,921,059 baz çiftleri arası
Bu Genin Diğer İsimlendirmeleri
- MGC70799
- MGC132008
- DPYD_HUMAN
- DPD
- DHPDHASE
- DHP
- dihidroürasil dehidrojenaz
- dihidrotimin dehidrojenaz
- dihidropirimidin dehidrojenaz [NADP +]
Kaynak: https://ghr.nlm.nih.gov/gene/DPYD
Görsel Kaynak: https://www.rcsb.org/3d-view/ngl/1gt8
Editör: Meryem GÖKOĞLU
