

İçindekiler
Tanım
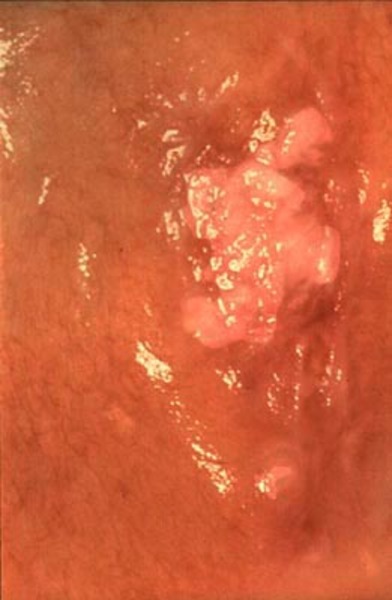
Diskeratozis Konjenita: Diskeratozis Konjenita, vücudun birçok bölümünü etkileyebilen bir hastalıktır. Bu hastalık üç özellik ile karakterizedir: el ve ayak tırnaklarında yetersiz ve anormal şekilde büyüme; genellikle “dantelli” olarak tanımlanan bir desende, özellikle boyun ve göğüs kafesindeki cilt renginde değişmeler (pigmentasyon), ağız içerisinde beyaz lekeler (oral lökoplaki) (Görsel 1).
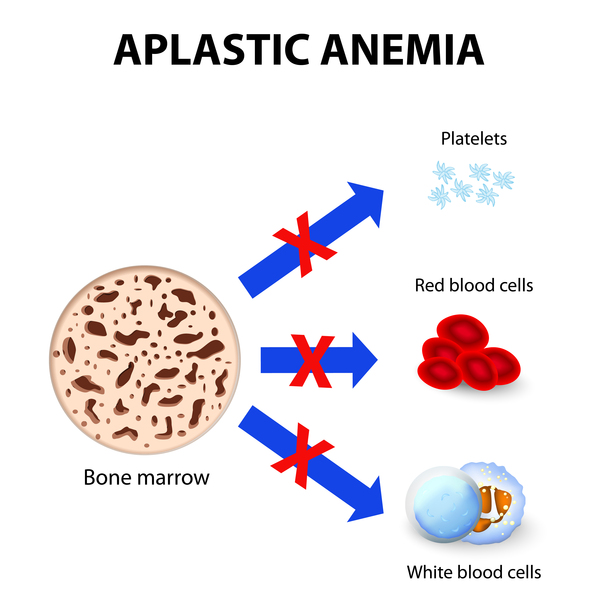
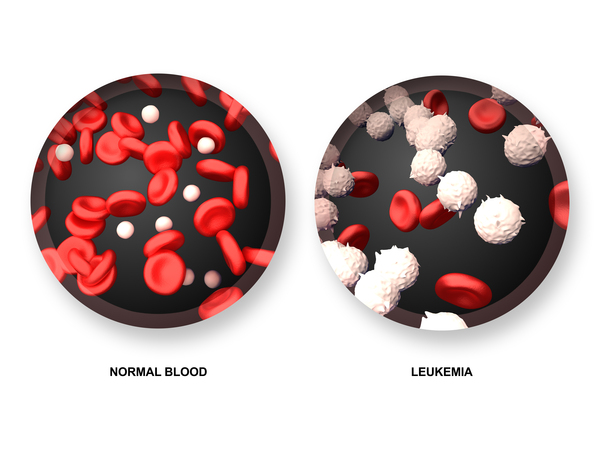
Diskeratozis konjenitaya sahip insanlar hayati tehlike oluşturan birkaç durum geliştirmede artan bir riske sahiptir. Bu insanlar, özellikle kemik iliği fonksiyonunu bozan rahatsızlıkla karşı korunmasızdırlar. Bu rahatsızlıklar kemik iliğinin yeni kan hücreleri üretme yeteneğini bozar. Etkilenen bireyler, kemik iliğinin yeteri kadar yeni kan hücresi üretmediğinde meydana gelen, ayrıca kemik iliği yetmezliği olarak da bilinen aplastik anemi (Görsel 2) geliştirebilirler. Olgunlaşmamış kan hücrelerinin normal olarak gelişemediği miyelodisplastik sendrom için ortalama riskten daha yüksektir; bu durum lösemi (Görsel 3) olarak adlandırılan bir kan kanseri formuna dönüşebilir. Hiç miyelodisplastik sendromu geliştirmeyen insanlar bile lösemi geliştirme riski altındadır. Ek olarak, özellikle baş, boyun, anüs ve cinsel organ kanserlerini geliştirmede diğer kanserlerden daha fazla ortalama riske sahiptirler.
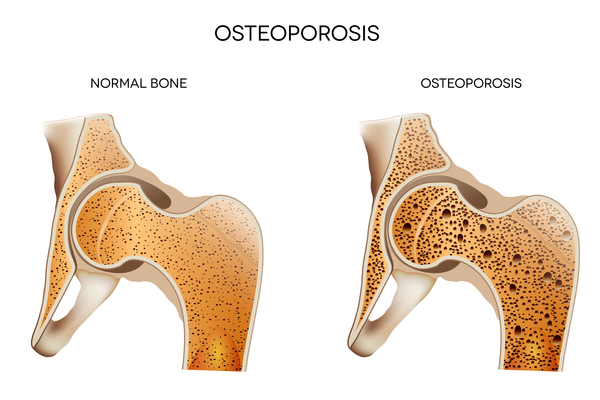
Diskeratozis konjenitaya sahip olan bireylerde ayrıca, ciğerlerde yara dokusunun (fibröz) gelişimine ve kan dolaşımında oksijen taşınmasını azaltan bir durum olan pulmoner fibroz geliştirilebilir. Diskeratozis konjenitaya sahip olan bazı insanlarda, kapalı olabilecek dar göz yaşı kanalları gibi göz anormallikleri, göz yaşının akmasını engellemesi ve göz kapağı tahrişine yol açması; diş problemleri; saç kaybı veya zamanından önce gri saç; düşük kemik mineral yoğunluğu (osteoporoz) (Görsel 4); kalça ve omuz eklemlerine bozulma (avasküler kangren); ya da karaciğer hastalığı gibi ek belirti ve semptomlar meydana gelir. Bazı etkilenen erkek bireylerde, idrarı mesaneden vücut dışına taşıyan tüp olan üretra (stenozu) daralması olabilir. Üretral darlık, zor ve acı verici idrar yapma ve idrar yolu enfeksiyonlarına neden olabilir.
Diskeratozis konjenitanın şiddeti bireyler arasında büyük ölçüde çeşitlilik gösterir. En az şiddetle etkilenen bireyler, normal kemik iliği fonksiyonuna ve hastalığın sadece birkaç hafif fiziksel özelliğine sahiptir. Daha şiddetli etkilenen bireyler, birçok karakteristik fiziksel özelliğe sahiptir ve kemik iliği yetmezliği, kanser ya da erken yetişkinlikte pulmoner fibroz deneyimlemiştir.
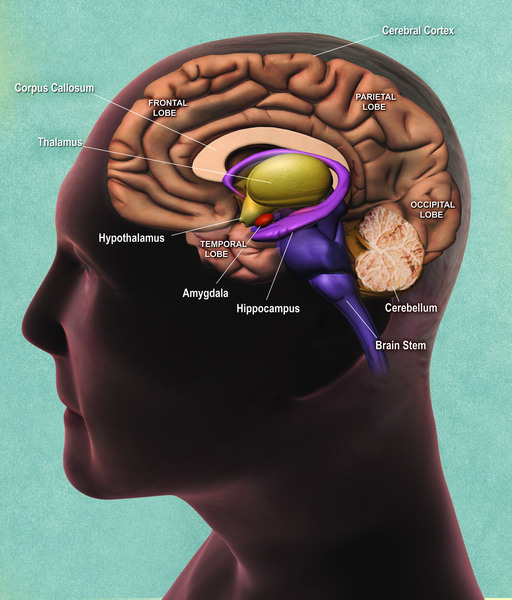
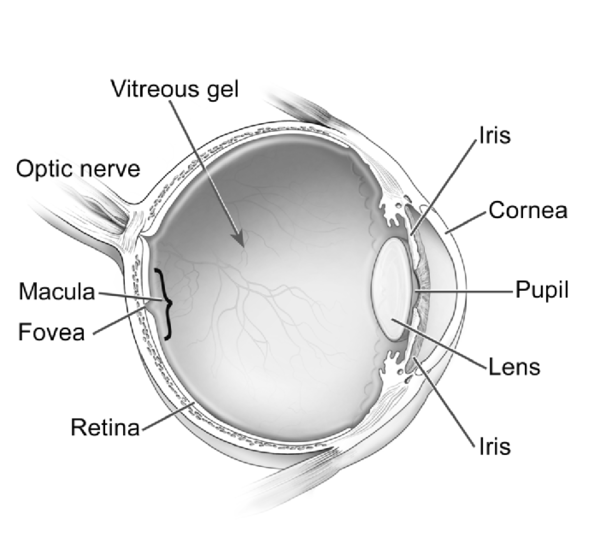
Diskeratozis konjenitaya sahip olan çoğu birey normal zeka ve ayakta durma ve yürüme gibi motor becerilerin gelişimine sahipken şiddetli bir şekilde etkilenen bazı bireylerde gelişimsel gecikme meydana gelebilir. Hastalığın şiddetli bir formu da Hoyeraal Hreidaarsson sendromu olarak adlandırılır, etkilenen bireyler, hareketi kontrol eden beynin bir parçası olan aşırı derecede küçük ve gelişmemiş beyinciğe (Görsel 5) sahiptirler. Revesz sendromu olarak adlandırılan diğer ciddi bir türü, diskeratozis konjenitaya ek olarak gözün arkasındaki (retina) (Görsel 6) ışığa duyarlı dokuda anormallikler içerir.
Sıklık
Diskeratozis konjenitanın kesin yaygınlığı bilinmemektedir. Yaklaşık olarak 1 milyon kişide 1 meydana geldiği tahmin edilmektedir.
Nedenler
Diskeratozis konjenitaya sahip olan insanların neredeyse yarısında hastalık, TERT, TERC, DKC1, ya da TINF2 genlerindeki mutasyonlardan kaynaklanır. Bu genler kromozomların uçlarında bulunan telomer (Görsel 7) adı verilen yapıların korunmasını sağlayan proteinlerin yapımı için talimatlar sağlar. Diskeratozis konjenita olan az sayıda bireyde telomer bakımıyla ilgili diğer genlerdeki mutasyonlar tanımlanmıştır. Diğer etkilenen bireyler şu an ki diskeratozis konjenita ile ilişkili hiçbir genlerinde mutasyona sahip değildir. Bu durumda, hastalığın nedeni bilinmemektedir ama yüksek ihtimalle telomer korunumu ile bağlantılı diğer tanımlanmayan genler söz konusudur.
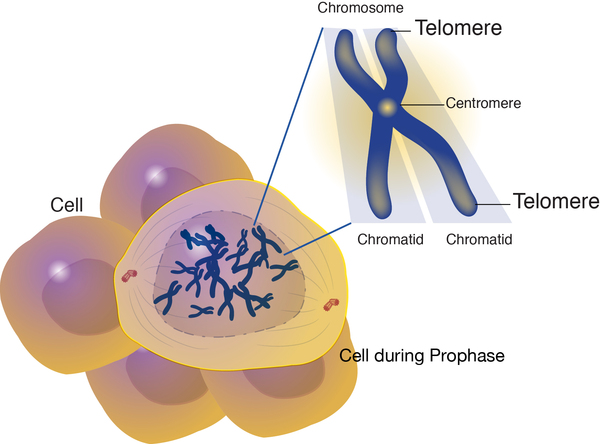
Telomerler, kromozomların anormal şekilde birbirlerine yapışmasından veya ayrılmasından korumaya yardımcı olur. Çoğu hücrede, telomerler hücre bölündükçe devamlı olarak kısalır. Belirli bir hücre bölünme sayısından sonra telomerler o kadar kısalırlar ki hücrenin bölümeyi durdurmasına ya da kendi kendini yok etmesine (apoptoza uğrar) neden olurlar.
Telomerler, telomeraz ve shelterin olarak adlandırılan iki önemli kompleks tarafından korunurlar. Telomeraz her bir hücre bölünmesinde kromozomların uçlarına tekrar eden küçük DNA parçaları ekleyerek normal telomer uzunluğunun korunmasına yardım eder. Telomerazın ana bileşeni hTR ve hTERT’ dir; bunlar yazılış sırasına göre TERC ve TERT genlerinden üretilir. HTR bileşeni DNA’ nın kimyasal bileşeni olan bir RNA molekülür. Telomerazın kromozomların uçlarına eklediği tekrarlanan DNA sekansını oluşturmak için bir şablon sağlar. HTERT bileşeninin fonksiyonu yeni DNA parçasını kromozomun uçlarına eklemektir. DKC1 geni telomeraz fonksiyonunda önemli başka bir proteini yapmak için talimatlar sağlar. Dyskerin olarak adlandırılan bu protein hTR’ ye bağlanır ve telomeraz kompleksinin sağlamlaştırılmasına yardım eder.
Shelterin kompleksi telomerleri hücrelerin DNA tamir işlemlerinden korunmasına yardımcı olur. Shelterinin koruması olmadan, tamir mekanizması DNA dizilimindeki anormal kırılmalarla kromozoma son verdiğini ve uçları birleştirmeye veya apoptozu başlatmaya çalıştığını algılardı. TINF2 geni shelterin kompleksinin parçası olan bir proteinin yapımı için talimatlar sağlar.
TERT, TERC, DKC1 veya TINF2 gen mutasyonları telomer uzunluğunun kısalmasını ve telomerlerin korunumunun bozulmasına neden olan telomer ya da shelterinin işlevsizliği ile sonuçlanır. Hızlı bir şekilde bölünen hücreler kısalan telomerlerin etkilerine karşı özellikle savunmasızdırlar. Bunun bir sonucu olarak diskeratozis konjenitaya sahip olan insanlar, vücutta tırnak yatakları, saç kökü, deri, ağız astarı (oral mukoza) ve kemik iliği hücreleri gibi kolayca bölünebilen hücreleri etkileyen çeşitli problemler deneyimleyebilirler.
Yetersiz telomer korunumundan kaynaklanan kromozomların dengesizliği ve kırılması, kontrol edilemeyen şekilde hücre bölünmesine izin veren genetik değişikliklere neden olabilir, bu da diskeratozis konjenitaya sahip olan insanlarda kanser gelişimine neden olur.
Kalıtım Modeli
Diskeratozis konjenita farklı kalıtım modellerine sahip olabilir. Dyskeratosis congenita DKC1 genindeki bir mutasyondan kaynaklandığında, X bağlantılı çekinik bir model (Görsel 8) ile kalıtılır. DKC1 geni iki cinsiyet kromozomundan biri olan X kromozomunun üzerinde bulunur. Erkeklerde (sadece bir X kromozomu olan) her hücredeki genin bir değişmiş kopyası bu duruma sebep olmak için yeterlidir. Kadınlarda (iki tane X kromozomu olan) hastalığa sebep olmak için genin her iki kopyasında mutasyon gerçekleştirmesi gerekmektedir. Kadınların bu gendeki iki değişmiş kopyaya sahip olma ihtimali düşük olduğundan dolayı erkekler X’ e bağlı çekinik hastalıklardan kadınlara göre çok daha fazla etkilenirler. X’ e bağlı kalıtımın bir özelliği babalar oğullarına X’ e bağlı özellikleri aktaramazlar.

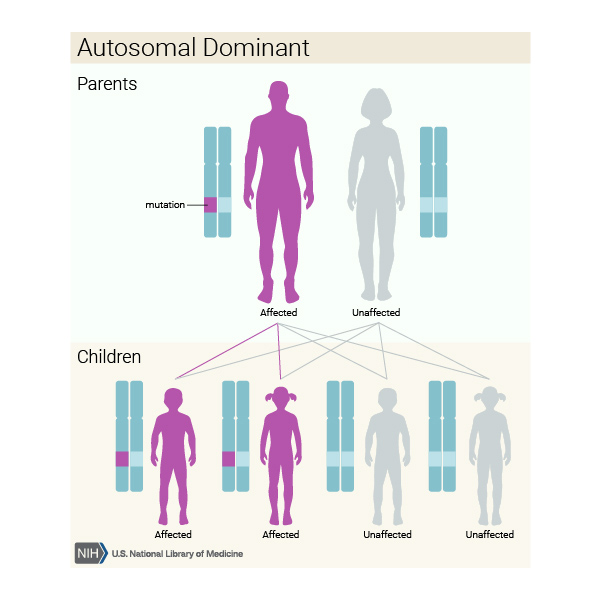
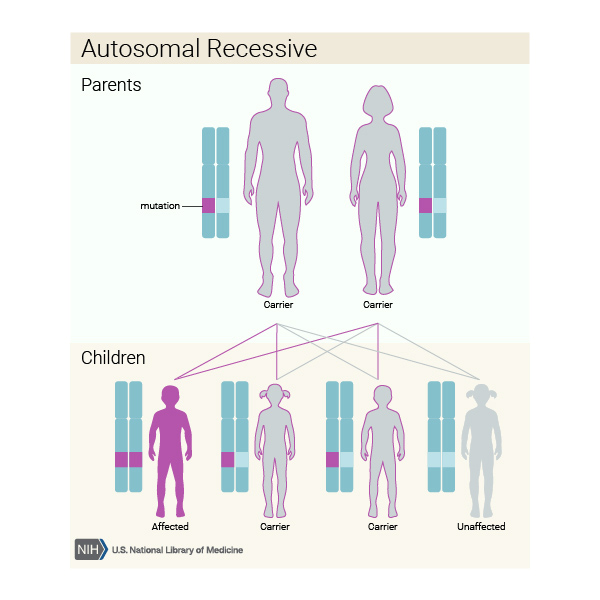
Diskeratozis konjenita başka genlerdeki mutasyonlardan kaynaklandığında, otozomal dominant Görsel 9) ya da otozomal resesif (Görsel 10) bir model ile kalıtılabilirler. Otozomal dominant, her bir hücrede değiştirilmiş genin bir kopyası hastalığa sebep olmak için yeterlidir demektir. Otozomal resesif her hücredeki genin iki kopyası da mutasyona sahip olduğu anlamına gelir. Otozomal resesif durumdaki bir bireyin anne ve babasının her biri mutasyonlu genin bir kopyasını taşır ama bu rahatsızlığın belirti ve semptomlarını göstermezler.
Bu Hastalığın Diğer İsimleri
- Zinsser-Cole-Engman sendromu
Kaynak: https://ghr.nlm.nih.gov/condition/dyskeratosis-congenita
Görsel Kaynak: https://www.ncbi.nlm.nih.gov/books/NBK22301/figure/dkc.F1/?report=objectonly
Editör: Meryem Melisa KAR