İçindekiler
Tanım
Megasistik-Mikrokolon-Intestinal Hipoperistaltizm Sendromu (MMIHS), idrar kesesini (1) ve bağırsakları (2) hizalayan kasları etkileyen şiddetli bir bozukluktur. Gıdaların sindirim sistemi boyunca peristalsis hareketi ile ilerlemesini sağlayan ve mesaneyi boşaltan kas kasılmalarının işlevini kaybetmesi ile karakterizedir.
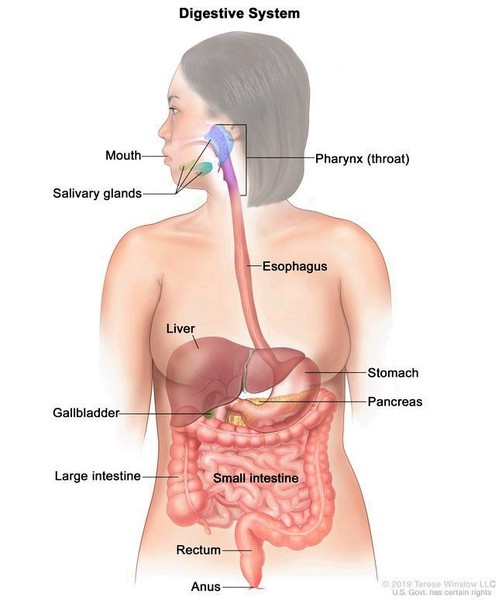
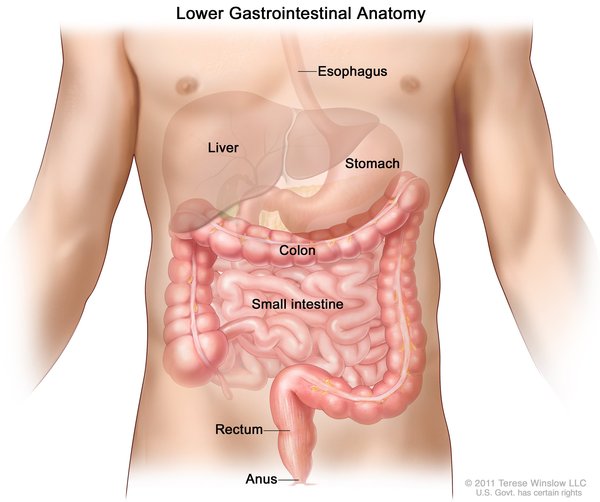
MMIHS’nin birkaç temel özellikleri doğumdan önce ultrason görüntüleme tekniği kullanılarak anlaşılabilir. Durumdan etkilenmiş fetüsler, boş olmadığı için genişlemiş bir mesaneye (megasistis) sahiptir. Ayrıca kalın bağırsak (kolon), onu kaplayan fonksiyonel kas eksikliği nedeniyle abnormal derecede dardır (mikrokolon). Bağırsak ve idrar kesesi problemleri hastaların yaşamı boyunca sürer.
Doğumun sonrası, peristaltizmin süren durumu (hipoperistalsis) genel olarak bağırsağın yalancı tıkanıklığı olarak da bilinen bir sindirim bozukluğuna sebep olur. Bağırsaklardaki fiziksel bir tıkanıklık gibi görünen fakat gerçek bir tıkanma olmayan bu durum, bağırsaklarda kısmen sindirilmiş yiyecek posalarının birikimine yol açar. Bu birikim, karnın şişmesine ve ağrımasına, mide bulantısına ve kusmaya yol açabilir. Kusmuk genellikle safra sıvısı adı verilen yeşil ya da sarı bir sindirim sıvısı içerir. Sindirim engellendiği ve vücut besinleri gıdalardan alamadığı için, genellikle intravenöz beslenmeyle (parenteral beslenme) verilen beslenme takviyesi yapılmalıdır. Etkilenen bazı hastalar sadece intravenöz beslenmeye güvenirken, diğerleri bunu sadece ara sıra gerektirir. Uzun süreli parenteral beslenme karaciğer problemlerini meydana getirebilir.
İdrarın vücuttan atılması kabiliyetinin azalması, karında ağrılı şişkinliğe de artırır. MMIHS’li birçok hasta, idrarı atabilmek için bir tüp (idrar sondası) kullanır. MMIHS’li bazı bireylerdeki bir başka semptom da , bağırsakların normal şekilde katlanmadığı bağırsak malrotasyonudur. Bunun yerine anormal bir şekilde bükülürler ve çoğunlukla tıkanmaya sebep olurlar. MMIHS’li hastalar, idrarı böbreklerden mesaneye taşıyan kanallar olan üreterlerle veya böbreklerle ilgili sorunlarla da karşılaşabilirler.
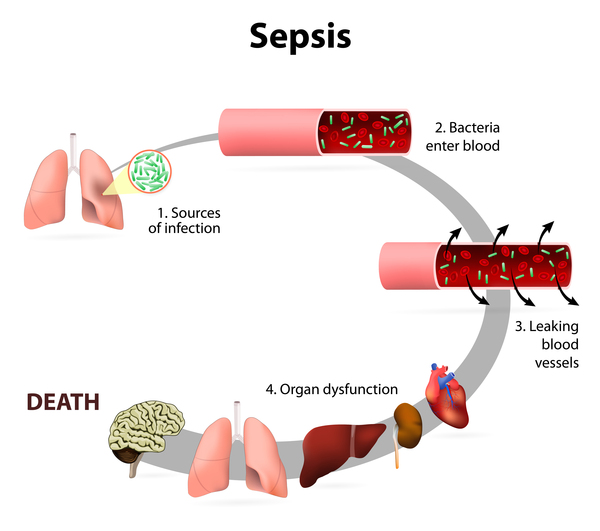
MMIHS’li hastaların beklenen yaşam süreleri, genellikle yetersiz ve dengesiz beslenme, ezici enfeksiyon (sepsis) (3) veya birçok organın yetmezliği gibi semptomlar ile normalden daha kısa sürmektedir.
Sıklık
MMIHS nadir bir hastalık olmakla beraber tıp literatüründe tanımlı 200’den fazla hasta bulunmaktadır.
Nedenler
MMIHS birden fazla gende meydana gelen mutasyonlardan kaynaklı olabilir fakat en çok çalışılan gen ACTG2 genidir. Bu gen gamma (γ)-2 aktin (4) adı verilen bir proteinin üretimi için gerekli emirleri verir. γ-2 aktin proteini ise kas liflerinin hareketini, kasılması ve özellikle idrar ve bağırsak yollarındaki düz kaslarının kasılması için önemli olan filamentler halinde düzenlenir. Bu kasılmalar, idrarın idrar kesesinden boşaltılmasını sağlar ve gıdaları bağırsaklardan geçirir.
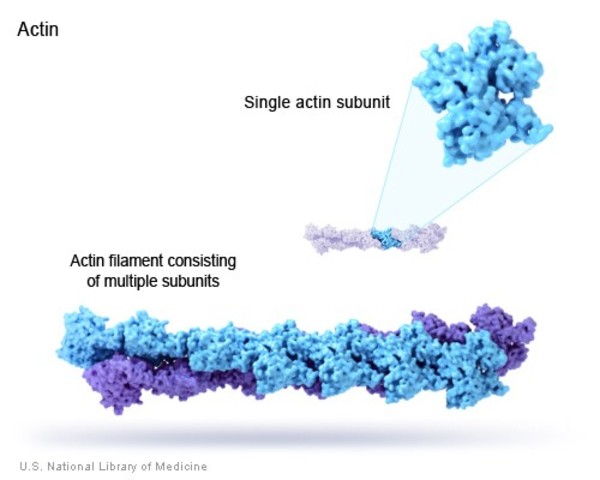
ACTG2 genindeki mutasyonlar, değiştirilmiş bir γ-2 aktin proteininin üretimine neden olur. Bu değişimler, mesane ve bağırsaklardaki düz kasların kasılma kabiliyetini bozan aktin filamentlerinin oluşumunu engeller. Kas kasılmaları ile alakalı bu problemler, idrarın boşaltılmasını ve yiyeceklerin bağırsaklardaki hareketini bozarak MMIHS’nin tipik sendromlarının görülmesine sebep olur.
Diğer genlerde oluşan mutasyonların ise nadiren MMIHS vakalarının sebebi olduğu bilinmektedir. Bu genler sayesinde üretilen proteinler aynı zamanda düz kas kasılmasında görevlidir. MMIHS’li kişilerin hemen hemen yüzde 10’unda tanımlanmış genlerinden birinde mutasyon yoktur. Tanımlanmamış diğer genlerin de duruma dahil olması olasıdır.
Kalıtım Modeli
Hastalık, ACTG2 genindeki mutasyonlardan kaynaklı olduğunda, otozomal dominant kalıtım modeline (5) sahiptir. Bunun anlamı her hücredeki değişmiş genin bir kopyası bozukluğa neden olmak için yeterli olabileceğidir. Bu tip durumlar, üreme hücreleri yani yumurta veya spermin oluşumunda ya da erken embriyonik gelişim döneminde oluşan gendeki yeni (de novo) mutasyonlardan kaynaklanır. Bu tür hastalıklarda, etkilenmiş bireylerin ailelerinde ya da akrabalarında hastalık geçmişi bulunmaz.

Durum, ACTG2 geni haricindeki diğer tanımlanmış genlerde olan mutasyonlardan kaynaklı olduğunda ise bozukluk otozomal dominant resesif modeline sahiptir. Bunun anlamı ise her hücrede bulunan genin ancak değiştirilmiş iki kopyasının hastalığa neden olabileceğidir. Otozomal resesif rahatsızlığı olan bir bireyin ebeveynleri mutasyona uğramış genin bir kopyasını taşırlar fakat tipik olarak durumun belirti ve semptomlarını göstermezler. Çünkü onlar sadece bu hastalığın taşıyıcısıdır.

Bu Hastalığın Diğer İsimleri
- Berdon sendromu
- Megacystis, mikrokolon, hipoperistalsis sendromu
- MMIH sendromu
- MMIHS
Görsel Kaynak: https://rottenpanda.com/diet-tips-and-home-remedies-for-appendicitis/
Editör: Züleyha DEMİRCİ